




皮肤正常的衰老过程不可避免,但世界护肤中心研究发现:85%的肌肤问题(如干燥、发黄、黯淡无光泽、松弛、皱纹)的元凶为盲目保养。美容基因检测使人们能够准确的了解自己的皮肤基因信息,从遗传因子的层面上找到皮肤问题的根源,剖析皮肤将面临的问题和人体衰老倾向,选择更适合自己的产品,结合科学的生活行为方式和美容护肤等手段预防皮肤问题,让护肤行为可以有的放矢。
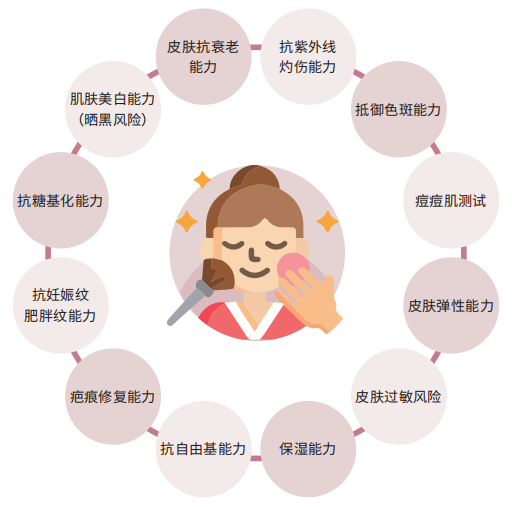
检测内容(12 项)
适用人群
■ 不知如何选择适合自己肤质的美容产品的人士
■ 美容 / 护肤品使用过多但皮肤问题依旧无法解决的人士
■ 想要更多了解自己为美丽健康储备信息的人士
样本类型
■ 血样采集卡
■ EDTA/ 枸橼酸钠抗凝血
■ 口腔粘膜上皮细胞


